Processing one-dimensional NMR data
1D-NMR data
One-dimensional NMR datasets are stored in a folder containing the measured data as an FID file (Free induction Decay) and separate files listing all measurement and processing parameters.
An NMR dataset in opened by simply dragging and dropping the data folder into Mnova. Automatically the FID data is converted into a spectrum by fourier transforming it. A fourier transform transforms the data from a time domain (time on the x-axis) into a frequency domain (frequency in the x-axis).
The first time you open a NMR data set you have to set the display properties. Right click in the spectral window and select 'Properties'
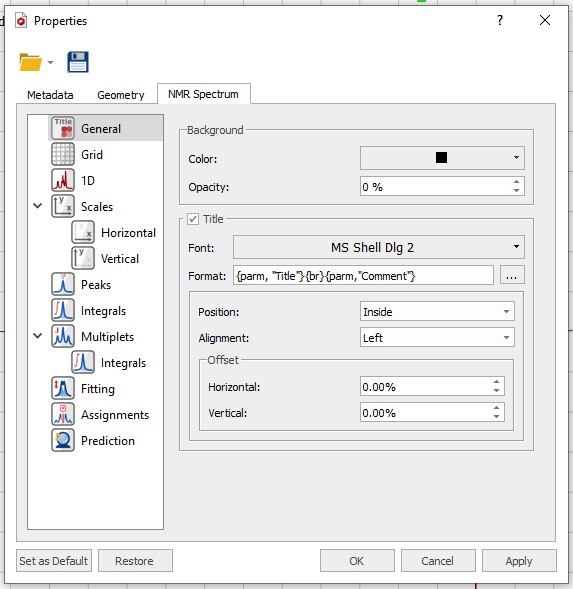
Download the NMR Spectrum Properties file located here and load in into MestreNova using the 'Folder' symbol at the top of the 'Properties' Window. After loading the file click the 'Apply' and 'Set as Default' buttons at the bottom of the 'Properties' window followed by 'OK'.
Before interpreting the spectrum you should execute the following manipulations:
Zoom in/out
While processing and interpreting your spectrum it is sometimes neccesary to zoom in to specific spectral regions or peaks. All magnification functions can be found in the 'View' ribbon
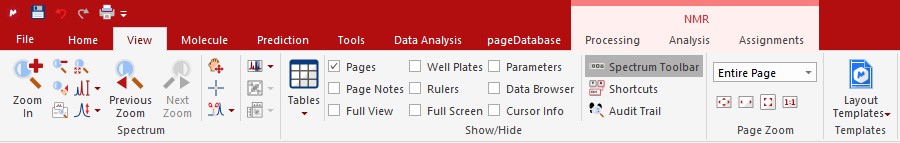
The zoom functions can also be accessed via keyboard short-cuts; 'Zoom In' (z), 'Zoom Out' (shift + z), 'Full Spectrum'(f). Besides the short-cuts, the 'Spectrum Toolbar' from the 'View' ribbon also adds the most used magnification buttons to the right hand side of the screen for easy access. In order to zoom in the vertial direction simply use the scroll wheel of your mouse.
Apodization
When the signal to noise ratio of a spectrum is poor one could off-course measure more scans, however this can be very time consuming and the available NMR-time per sample is limited. Another approach to increase the 'signal to noise ratio' is to multiply the FID by an exponential function before Fourier transforming. This increase in signal to noise is not without a penalty, by applying the exponential function the resolution of the spectrum will decrease. In the ‘Processing’ menu select the ‘Apodization’ option. In the pop-up menu you can choose between different window functions. Select the ‘Exponential’ function and choose an appropriate line broadening factor between 0 and ~1 Hz.
Phase Correction
In a high resolution NMR spectrum we want our peaks to be as narrow and symmetric as possible. By adjusting the phase of you can adjust the symmetry of the peaks and align the bottom of the peaks with the baseline. In the ‘Processing’ menu select ‘Manual Correction’. In the pop-up menu you can adjust ‘PH0’ and ‘PH1’, the zero and first order phase correction parameters. The biggest peak in the spectrum is automatically selected as pivot point. By clicking in the blue area and holding your left or right mouse button you can adjust ‘PH0’ and ‘PH1’ respectively by moving your mouse up or down. ‘PH0’ has effect over the whole spectral with, the effect of ‘PH1’ increases the further away from the pivot point.
Zero refencing
The spectrum shown is not yet referenced, which means that the signals you observe are not showing at the correct chemical shifts. To make sure that the spectrum is properly referenced the reference peak (TMS, or when no TMS is present in the sample the solvent residual peak) has to be set to the proper chemical shift.

To do so select the ‘Reference’ option in the ‘Analysis’ menu.
Position the red cursor line on the top of the reference peak and click, then enter the correct chemical shift in the pop-up menu.

Peak Picking
To annotate all interesting peaks select 'Manual Threshold’ from 'Peaks' group in the ‘Analysis’ ribbon or use the keyboar short-cut 'k'.
Now you can select different regions and height thresholds where peaks should be detected.
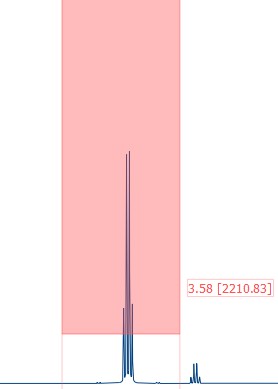
Single peaks can be picked using the 'Peak by Peak' button from the 'Peaks' group or using the keyboard short-cut 'Crtl + k'. The automatic peak picking option usually picks way to many points making your spectal image very crowded and unclear.
Peak Integration
To integrate peaks select ‘Manual’ from the 'Integrals' group in the ‘Analysis’ ribbon.
Select the regions to integrate by clicking and dragging.
The first selected region is always set to 1.00, to change this value, right click the integral value and select ‘Edit Integral’.


